The main ambition of our lab is to predict the regulatory evolution of gene expression and its causal association with trait evolvability.
Integrating comparative/functional genomics, evolutionary transcriptomics, and causal inference of gene regulatory networks into a predictive framework of trait evolvability
Linking genotype and phenotype to make predictive claims about trait evolvability and speciation has always been a grand aim of genetics, molecular biology and interfacing disciplines. While in the past much has been learned about individual molecular examples able to induce phenotypic or morphological change, a system-scale understanding of causal genotype-phenotype associations remains an open challenge. Recent advancements in computer science (HPC/cloud-computing, advances in machine learning and artificial intelligence), high-throughput sequencing technologies (massive genome sequencing and assembly efforts, massive sequencing read archives), and new methodologies in scientific software development (software containers, workflow/pipeline architectures) can now be combined to address this classic genotype-phenotype problem using data-driven methodologies at an unprecedented scale.
Learning from genomes at tree-of-life scale
On a daily basis, life scientists query genomic sequence data against reference genome databases to retrieve analytical insights about their genes or organism of interest. The search engine they use for this task, BLAST, is provided by the governmentally hosted National Center for Biotechnology Information (NCBI). For more than ten years the genomic search process remained the same in that life scientists spend tedious hours to manually evaluate search results from the NCBI HTML text output or at best semi-automated when dealing with the BLAST command line tool. Although the procedure is feasible for a small number of sequences (thousands), it remains impractical in its current form for large-scale genome searches at tree of life scale and when dealing with genes from millions of genomes. This creates an analytics bottleneck and slows down life science research and innovation. We intend to overcome this bottleneck by building a fundamentally novel sequence search infrastructure for large-scale sequence searches that enables life scientists and our own research to automate ultra-large sequence search requests and data interpretation at tree-of-life scale (Buchfink, Reuter, Drost, 2021).
The Earth BioGenome Project (Lewin et al., 2018) aims to assemble representative genomes for ~1.5 million eukaryotic species within the next decade and so far consists of individual community driven bulk-sequencing efforts (Exposito-Alonso, Drost et al., 2019). Such a wealth of genomic resources enables us to infer biological principles at an unprecedented scale and will allow us to reveal a vast diversity of biological strategies that evolved to generate adaptive traits.
We are contributing to the Earth BioGenome Project as part of the European Reference Genome Atlas (ERGA) and built a sensitive tree-of-life scale protein alignment infrastructure based on the protein aligner DIAMOND (Buchfink, Reuter, Drost, 2021) to enable and pioneer modern genomics research fuelled by millions of genomes. DIAMOND matches the alignment sensitivity of the gold-standard tool BLASTP when running in –very-sensitive and –ultra-sensitive modes, while achieving up to 360x computational speed-up. At the same time, sequence similarity >50% identity can still be sensitively retrieved with computational speedup against BLAST up to 8,000x.
We use this new sequence search potential for tree-of-life scale comparative genomics and functional genomics analyses and combine this genomic information with high-throughput sequencing data such as bulk- or single-cell RNAseq to make use of the evolutionary transcriptomics methodology (Drost et al., 2016b; Drost et al., 2018a).
Evolutionary Transcriptomics
Evolutionary transcriptomics studies combine insights from comparative genomics and high-throughput transcriptomics to screen in silico for the potential existence of evolutionary constraints within a biological process of interest (Drost et al., 2018a). This is achieved by quantifying transcriptome conservation patterns and their underlying gene sets in biological processes.
Applying evolutionary transcriptomics to study heterochronic shifts in development
For centuries, research focused on quantifying and comparing the diversity of morphological traits among diverse species to study the evolutionary and developmental processes that generate organismal complexity. This concept of characterizing common traits among phylogenetically related species to classify the organizational form of multicellular organisms during comparable stages of development is today referred to as body plan concept. This concept aims to provide a comparative framework to study the origin, evolution, and variability of organismal forms by relating the function of homologous traits between extant species. Today, transcriptomes allow us to retrieve insights about the molecular processes that govern the developmental series towards the establishment of a variety of morphological traits. This is achieved by determining stages of development that are more conserved in terms of gene expression patterns or in terms of homology confirmed by genic sequence identity that are most active at this stage (find more details here) (Drost, 2016c).
A prominent application of this technique is the developmental hourglass phenomenon initially observed in animal embryology, where embryos of diverse vertebrates share common anatomical features and express more homologous genes during the middle of embryonic development. The origin of this phenomenon has been linked to the Cambrian Explosion more than 500 million years ago where most anatomical features of animals are believed to have emerged within a short 20-40 million year time window (see a comprehensive summary here). Due to the limited time to evolve anatomical diversity, today’s animals are still developing variations of these morphological features that emerged during the Cambrian Explosion (however this notion is intensely debated). Since these features are developed in mid-embryo development, the anatomical similarity between vertebrate species might be a consequence of a constrained body plan (= conserved anatomical features shared across animals) that formed more than 500 million years ago.

A web of complex interactions among developmental modules results in body plan constraints during mid-embryogenesis in animals. The waist of the hourglass denotes the phylotypic period where modular interactions maximize and morphological divergence minimizes resulting in the bottleneck of the developmental hourglass model.
In collaboration with the Grosse Lab and Quint Lab, we tested the developmental hourglass hypothesis by studying the developmental transcriptome of plant embryogenesis. In contrast to animals, plants are the second major branch of eukaryotic life that evolved embryo development as a process to organize complex multicellularity. The last common ancestor of animals and plants, however, is roughly estimated to have lived ~1.6 billion years ago and was likely unicellular (Meyerowitz, 2002). As a result, multicellularity and embryo development must have evolved convergently in plant and animal lineages (Quint, Drost et al., 2012).

Using the model plant Arabidopsis and the software tools orthologr and myTAI developed by our lab, we studied the developmental transcriptomes covering plant embryonic- and post-embryonic development. As a result, we found that the transcriptomes of developing plant embryos and post-embryonic development follow transcriptomic hourglass patterns analogous to the transcriptomic patterns found in animal embryos (Quint, Drost et al., 2012; Drost et al., 2015; Drost et al. 2016a).
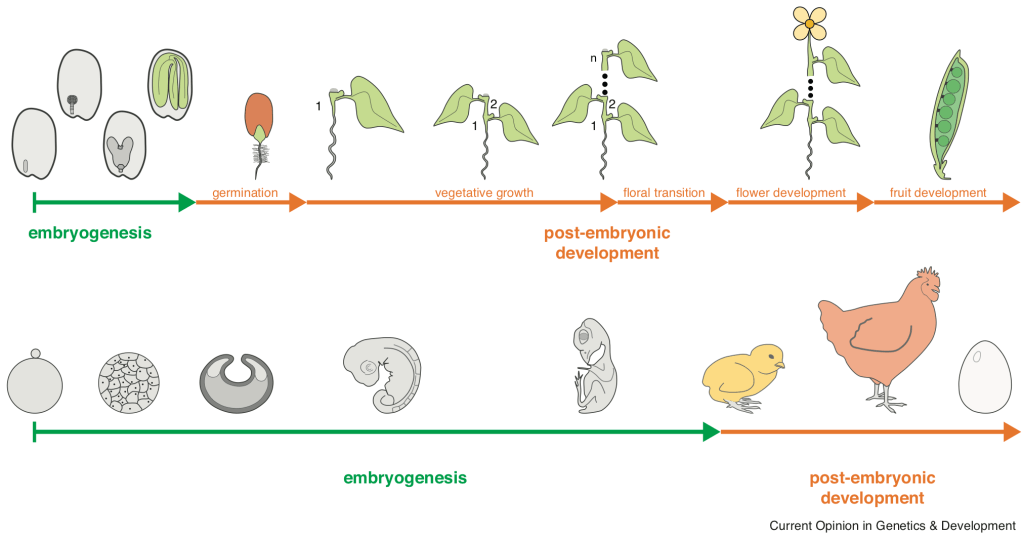
Comparing the life cycles of animals and plants. In most animals development is largely embryonic. The vast majority of organs are practically simultaneously initiated during the phylotypic period and body plan complexity of mature embryos is comparable to adults. In plants most organs develop post-embryonically. Mature embryos therefore only possess a basic body plan and are, consequently, less complex compared to adult plants.
The presence of a plant hourglass pattern suggests that both phenomena evolved independently in animals and plants and challenges the current hypothesis in animals that postulates that the origin of the morphological hourglass phenomenon is caused by constraints on body plan establishment alone (Quint, Drost et al., 2012; Drost et al., 2015; Drost et al. 2016a; Drost et al., 2017a; Drost et al., 2018a). It furthermore challenges the current notion that transcriptome conservation and morphological trait conservation in the developmental hourglass model have to be causally linked (Drost et al., 2017a). As an alternative model, we postulated the organizational checkpoint model which integrates the developmental hourglass model into a framework of transcriptome switches that convergently evolved in both lineages and which resemble a more fundamental process underlying (and possibly constraining) body plan diversification in both, plants and animals and potentially even fungi fruit body development (Drost et al., 2017a).

The presence of transcriptomic hourglass patterns in animals has been extensively debated in the past years. This figure summarizes the species for which transcriptomic hourglass patterns have been described. Plants and fungi are still underrepresented and further research is required to fill these gaps in species sampling.
We now study, whether organizational checkpoints mark stages in biological processes in which the sequential order of biological events (e.g. developmental programs) is maximally conserved due to the evolutionary constraints acting on key regulators or developmental modules within their system-scale gene regulatory network. The conservation of expression patterns of these developmental regulators is crucial to maintain the sequential order of development. Hence, environmental factors or genetic factors such as point mutations, recombination, or transposable elements able to cause the change of expression patterns of these regulators might result in the disruption of a developmental series and may as a consequence lead to heterochronic shifts in development which in turn are negatively selected due to constraints on the underlying organizational checkpoints.
Unveiling the transcriptional changes associated with the morphological diversity of plant organs
In collaboration with Christoph Schuster, the Meyerowitz Lab, the Leyser Lab, and the Grosse Lab, we harvested nine organs during crucial developmental stages of seven prominent plant model organisms and quantified their underlying transcript abundance using bulk RNAseq. As a result, we could confirm already known genes, splice variants, natural antisense transcripts (NATs), long-intergenic RNAs (linc RNAs), circular-RNAs (circRNAs) and other non-coding transcripts, but also identified yet unknown loci that can now be characterised according to the aforementioned functional categories. Due to the fine-grained phylogenetic distances between these seven flowering plant species (up to 150 Mya), we also quantified the rate of expression divergence of different organs to investigate the relationships between molecular divergence and morphological divergence in plants. Together, these resources aim to serve as a community reference for developmental and evolutionary studies and unravel key molecular players underlying morphological trait variation of plant organs.
Causal Inference of Gene Regulatory Networks
The developmental sequence common to animal embryogenesis across different species is: Fertilization –> Cleavage –> Gastrulation –> Neurolation and thus has a particular sequential order that evolved over evolutionary time. Changes in gene expression of key regulators governing these transitions might, therefore, disrupt the correct sequence of these developmental programs and might lead to malfunctions or the death of the embryo. This example shall illustrate how the sequence of developmental programs itself (once established during evolution) is constraining diversification (Dollo’s law or Riedl’s concept of burden). The reason why animals did not evolve any other sequence of embryonic developmental programs might be that any change in this sequential order has generated disadvantageous morphologies leading to the abortion of the embryo. Plant development is more plastic than animal development, but developmental instructions following a particular order are still crucial for establishing a species-specific morphology or adaptive trait (Drost, 2016c).
Our group seeks to understand how gene regulatory interactions evolve on the system level to give rise to either conserved or divergent subnetworks (= modules or pathways). Such modules can be involved in conserving developmental instructions or allowing for the emergence of novel adaptive traits. For this purpose, we make use of gene regulatory networks which denote a modeling concept to quantify gene regulatory changes over evolutionary time. In a gene regulatory network, nodes denote either genes or other regulatory agents and edges reflect either inferred or experimentally validated regulatory interactions between genes.
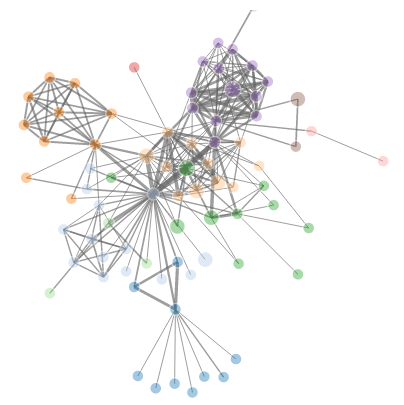
Using the comparative method, we can infer system-scale gene regulatory networks (sysGRNs) from transcriptomic data (taking gene ontology information into account) of different cell-types, ecotypes, or species and can detect modules either shared across different cells, ecotypes, or species or modules that are cell-type, ecotype, or species-specific (Moutsopoulos et al., 2021).
In collaboration with the Mohorianu Lab, our group builds the mathematical, computational, and comparative frameworks to quantify module retention and adaptive change in sysGRNs and relies on in-house generated as well as publicly available transcriptome datasets to infer such networks covering various biological processes at various resolutions of organismal complexity. Together, this network approach will unveil modules of developmental instructions that are associated with common morphological features shared between related species and provides a new analytical tool to screen for candidate pathways driving adaptive change in various environmental conditions.
Genome Dynamics and Transposable Elements
Understanding the contribution of transposable elements to genome dynamics and natural variation
In collaboration with the Paszkowski Lab, Sanchez Lab, Zabet Lab, and Catoni Lab, we studied how epigenetically controlled transposable elements contribute to changes in genomic regulation. From this research, we have gained fundamental insights into the biology of transposable elements and their influence on genome plasticity and genome evolution (Drost et al., 2017b; Sanchez et al., 2017; Gaubert et al., 2017; Cho et al., 2019; Benoit, Drost et al., 2019; Drost and Sanchez, 2019; Drost, 2020; Benoit and Drost, 2021; Srikant and Drost, 2021).
In particular, we demonstrated that the heat-responsive retrotransposon ONSEN is able to perform template switches during the extrachromosomal reverse transcription step within the retrotransposon lifecycle (Sanchez et al., 2017; Gaubert et al., 2017, Drost and Sanchez, 2019).

This finding challenges the current assumptions of retrotransposon evolution which do not take extrachromosomal retrotransposon recombination into consideration. This extrachromosomal recombination capacity of retrotransposons follows similar principles found in retroviruses and consequently enables retrotransposons to “rejuvenate” their sequence through extrachromosomal recombination between old and young family members with substantial consequences for the evolution of retrotransposon populations and host evolution. In this context, we introduced the notion of a selfish clan which denotes retrotransposon family members capable of activation and generation of mosaic progenies through inter-element recombination (Drost and Sanchez, 2019).
In a collaboration with Matthias Benoit, the Paszkowski Lab, the Catoni Lab, and Baulcombe Lab, we studied retrotransposon activity in crop plants to understand whether specific transposon families can be activated artificially and harnessed to speed-breed crops for selective plant breeding in response to global warming. For this purpose, we characterised the abundance and epigenetic control of the retrotransposon family RIDER in tomatoes which has previously been shown to be involved in tomato fruit shape elongation. This was achieved by de novo re-annotating functional retrotransposons in >110 plant genomes using LTRpred: a functional retrotransposon annotation pipeline developed in our lab (Drost, 2020).

Summary and individual steps of the LTRpred functional retrotransposon annotation pipeline. The LTRpred pipeline can be divided into four major modules: Preprocessing, De Novo Annotation, Postprocessing, and Filtering Analysis (Drost, 2020).
Further wet-lab experiments and characterization of RIDER within the plant kingdom (using metablastr; a software tool developed in our lab) revealed evidence that RIDER elements can be activated via drought stress and may help plants rich in RIDER activity to better adapt to drought stress conditions (Benoit, Drost et al., 2019). We also annotated another retrotransposon family, FIRE, which is a retrotransposon that belongs to the Gypsy superfamily. FIRE has recently been identified as active in the pericarp tissue of mature tomato fruits, likely as a consequence of reduced DNA methylation. So far, it was unknown whether FIRE is present in other plant species and whether its evolutionary trajectory can be traced across the plant kingdom. Our analysis revealed 196 BLAST hits, sharing at least 60% sequence homology with the FIRE reference, distributed between three Solanaceae species: Solanum lycopersicum, Solanum pennellii and Solanum habrochaites indicating extensive natural variation within the FIRE population in Solanum pennellii and Solanum habrochaites, in which full-length FIRE elements are not found, and the dominant FIRE population contains elements sharing approx. 80% sequence homology over 7500 bp of the FIRE reference, as seen in Solanum lycopersicum (Benoit & Drost, 2021).

In collaboration with the Cho lab, we contributed to the development of a novel technique named ALE-Seq which is a molecular biology technique to identify and quantify extrachromosomal linear (ecl) DNA derived from expressed genomic loci (Cho et al., 2019). ALE-Seq allows to determine the mobilization trigger of young and structurally intact elements that were initially annotated with LTRpred and thus enables classification of mobile retrotransposons families according to their environmental stimulus (= activation annotation). Using this technique we identified three active retrotransposon families in tomato and rice (RIDER, FIRE, Go-On) and provided a comprehensive pipeline from wet-lab to data analytics able to annotate mobile retrotransposons in a diverse range of species. This pipeline will be specifically useful for characterizing mobile retrotransposons in crops and will subsequently allow to engineer artificial retrotransposon mobilization for targeted crop breeding.
